|
||||
|
Глава V Плоть от плоти моей, кость от кости моей [о скелете] 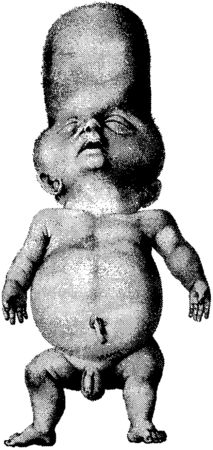
Около 1896 года на мысе Доброй Надежды появился китайский матрос по имени Арнольд. Нам мало что о нем известно, и портреты его до наших дней не дошли. Однако мы можем предположить, что он был невысок ростом, с сильно выступающим лбом. Еще он был слаб на голову – не в смысле интеллекта, а потому что верхние кости черепа у него отсутствовали. Вероятно, у него не было ключиц, а если и были, то не доставали до лопаток. И если бы кто-то, стоящий сзади, толкнул его, плечи Арнольда могли бы легко сложиться, как книжка, перед грудью. У него, возможно, были лишние зубы, а может, их и вовсе не было. Мы можем высказать столько догадок потому, что Арнольд был исключительно плодовитым и многие из его многочисленных потомков обладают вышеописанными чертами. Прибыв в Кейптаун, он обратился в мусульманство, завел семерых жен и погрузился в жизнь капско-малайского общества. Капские малайцы – это люди в основном яванского происхождения, вобравшие в свой генетический состав примесь народов сан, кхой, западных африканцев и мальгашей. Традиционно занимающиеся ремеслами и рыболовством, капские малайцы создали элегантные фронтоны особняков в стиле Капская Голландия (Кейп-Датч) для владельцев южноафриканских винодельческих поместий, внесли в национальную кухню специфический восточный аромат и обогатили язык африкаанс небольшим количеством малайских слов наподобие "пайсанг"[122]. Проведенное в 1953 году обследование выявило мутацию отсутствия костей у 253 потомков Арнольда. В 1996 году мутация распространилась среди тысячи человек. К счастью, отсутствие ключиц и местами мягковатый череп не представляют большой опасности для здоровья. Клан Арнольда и в самом деле вполне справедливо гордится своим прародителем и дарованной им мутацией.[123] Формирование костей Возможно потому, что кости последними из наших бренных останков обращаются в прах, мы думаем о них как о чем-то неживом. Но они таковыми не являются. Подобно сердцу и легким, кости постоянно формируются и разрушаются, как того и требует цикл созидания и деструкции. И хотя они кажутся такими отличными от остального тела, они происходят из тех же самых эмбриональных тканей, которые создают покрывающую их плоть. В самом полном смысле слова можно сказать, что кость – это трансформированная плоть. Тесная взаимосвязь между костью и плотью может быть продемонстрирована на примере происхождения образующих их клеток. Большая часть костных клеток – остеобластов – берет свое начало в мезодерме, той же самой зародышевой ткани, из которой формируются также соединительная ткань и мышцы. Взаимосвязь проявляется и в том, как образуются кости. Глубоко внутри каждой кости сохраняются остатки создавших ее клеток. Различные кости нашего организма формируются двумя разными способами. Плоские кости, такие как кости черепа, начинают складываться у зародыша как слои остеобластов, секретирующих белковую основу (матрикс). На этой основе образуются зубчики фосфата кальция, которые заключают клетки в оболочку. По мере роста кости добавляются новые слои остеобластов, каждый из которых в свою очередь инкапсулируется за счет собственной секреции. У длинных костей наподобие бедренной процесс идет несколько по-иному. Их формирование начинается с конденсаций клеток, заметных в развивающихся конечностях зародыша. Эти клетки, также производные мезодермы, называются хондроцитами, и они продуцируют хрящевую ткань. Хрящ – это шаблон для будущей кости, который лишь позднее станет добычей остеобластов. При своем первом появлении шаблон похож на кость своей формой, но не структурой. Одна из молекул, контролирующая клеточные конденсаты, – это костный морфогенетический белок (bone morphogenetic protein – BMP). Для удобства можно говорить о нем как об одной молекуле, хотя в действительности их целая семья. Подобно многим другим семействам сигнальных молекул, BMP обнаруживаются в самых неожиданных местах зародыша. Именно BMP задолго до формирования костей приказывают некоторым зародышевым клеткам становиться животом, а не спиной. Однако на более поздних стадиях развития эмбриона BMP появляются в скоплениях клеток, которые в будущем станут костями. У детей и взрослых они возникают в местах переломов костей. Поразительное свойство BMP состоит в их способности вызывать индукцию кости практически повсюду. Если сделать крысе подкожную инъекцию BMP, у нее сформируются костные разрастания совершенно отдельно от скелета, но со структурой обычной кости, вплоть до наличия костного мозга. Для создания кости недостаточно, чтобы недифференцированные клетки сконцентрировались в нужном месте и в нужном количестве. Клетки должны превратиться в остеобласты и хондроциты. Возвращаясь к метафоре, которую я уже употреблял ранее, они должны скалькулировать свою судьбу. Ген, отвечающий за судьбу остеобластов, тот же самый, что вызвал появление синдрома "головы Арнольда".[124] Этот ген кодирует транскрипционный фактор под названием CBFA1. Можно подумать, что CBFA1 не имеет большого значения, поскольку в результате его мутации обнаруживается недостаток всего лишь нескольких костей. Однако все потомки Арнольда гетерозиготны по этой мутации: только один из двух генов CBFA1 является носителем мутантной копии. Мыши, гетерозиготные по мутации того же гена, также отличаются неокостеневшими головками и отсутствием ключиц. Но мыши, гомозиготные по мутантному гену, практически не имеют костей. Вместо скелета у них имеются лишь хрящевые тяжи, пронизывающие весь организм, а мозг у них защищен всего только слоем кожи. Они могут сгибаться во всех направлениях, но это им не нужно, так как они мертвые. Бескостные мыши умирают в течение нескольких минут после рождения от удушья из-за того, что у них нет грудной клетки для поддержки легких.[125] По одной из странных причуд истории генетики Южная Африка является также родиной мутации, которая по своему действию противоположна той, что возникла у Арнольда, так как вызывает не дефицит костного вещества, а, напротив, его избыток. Вместо дыр из-за отсутствия костей в черепе жертвы этой второй мутации обладают крайне массивными черепами. При рождении мутация никак себя не обнаруживает. Толстые кости черепа и грубые черты лица, являющиеся характерными для этого синдрома, развиваются только с возрастом. В отличие от мутации утраты костей, мутация избытка костной ткани нередко является летальной. Ее носители обычно умирают в зрелом возрасте от судорог, так как дополнительные костные образования пережимают некоторые жизненно важные нервы. Опять-таки, в отличие от мутации отсутствия костей, мутация толстых костей черепа рецессивна и выражена лишь у небольшой горстки людей, жителей одной деревни, которые заключают браки между собой и ведут свое происхождение от одного голландца, основавшего в XVII веке поселение Капская колония. Вызывающая это нарушение мутация выводит из строя совершенно другой, чем CBFA1, ген. Сам белок носит название склеростина вслед за вызываемым им синдромом – склеростозом.[126] Считается, что он является ингибитором молекул BMP – возможно, связывая их, а потом выводя из строя. Именно так действуют многие ингибиторы BMP. На ранних стадиях зародышевого развития молекулы организатора наподобие ноггина ограничивают действие BMP как раз таким способом. В самом деле, мутации ноггина приводят к еще одному синдрому избыточного роста костной ткани, поражающему только пальцы рук и заставляющему их с возрастом срастаться вместе, делая их неподвижными.[127] Нарушения, связанные с избытком кости, иллюстрируют необходимость того, что организм должен держать семейство BMP под контролем. И все же сросшиеся пальцы и даже толстые кости черепа – это относительно слабые проявления способности BMP продуцировать костную ткань в самых неподходящих местах. Есть еще одно заболевание, демонстрирующее степень нарушений, которые могут произойти, если остеобласты распространятся по всему организму и начнут образовывать кости где им вздумается. Этот недуг называется прогрессирующей окостеняющей фибродисплазией (fibrodisplasia ossificans progressive), или ПОФ. Это редкое заболевание: по подсчетам, число людей, страдающих им во всем мире, составляет около 2500, но специалисты в этой области наверняка знают о существовании лишь нескольких сотен больных. Самой известной жертвой недуга был американец по имени Гарри Реймонд Истлэк. 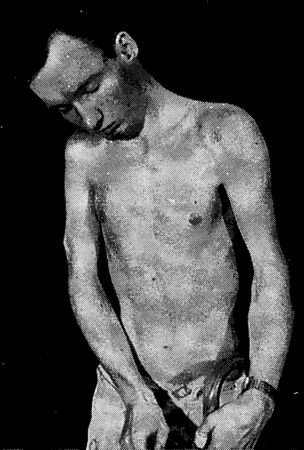
В 1935 году Гарри, которому тогда исполнилось пять лет, сломал ногу, играя с сестрой. Перелом сросся неудачно, и у мальчика навсегда осталась искривленная левая бедренная кость. Вскоре после этого случая у него также выявилось нарушение подвижности в тазобедренном и коленном суставах, что, однако, не было следствием первоначального перелома, а вызывалось костными отложениями, сформировавшимися на отводящей и четырехглавой мышцах. Гарри рос, а костные образования распространялись по всему телу. Они появились в ягодицах, на груди и шее, а также на спине. К 1946 году его левая нога и тазобедренный сустав полностью окостенели; туловище приобрело постоянный наклон под углом 30°; костные соединения образовали мост вдоль всего позвоночника, а мышцы на спине превратились в костные пластины. Делались попытки удалять эти образования хирургическим путем, но они отрастали снова и становились еще тверже и обширней. В возрасте двадцати трех лет он был помещен в специальную клинику для хронических больных. Он умер в 1973 году. К этому времени у него срослись челюсти, и он уже не мог разговаривать. По просьбе Гарри Истлэка его скелет передали для научных исследований, и теперь он стоит в витрине Мюттеровского музея в Филадельфии. Изобилующий дополнительными костными тяжами, распорками и выростами, которые оплели конечности и грудную клетку, его скелет выглядит так, как будто обычный костяк сорокалетнего человека поместили внутрь другого скелета, формирование которого уже началось, но вышло из-под контроля. Причина болезни в общих чертах понятна. Травмы тканей вызывают у больных ПОФ ненормальную реакцию. Вместо того чтобы лечить синяки и растяжения с помощью соответствующих тканей, в ход идут остеобласты и образовавшиеся участки превращаются в кость. Это типичные проявления ошибок в продукции BMP или контроле над ними, но причина этих сбоев, т.е. сама мутация, до сих пор не обнаружена. Поиски ее могут оказаться долгими. У больных ПОФ редко рождаются дети, так что ген, являющийся первопричиной заболевания, нельзя определить путем изучения длинных родословных пораженных недугом семейств.[128] 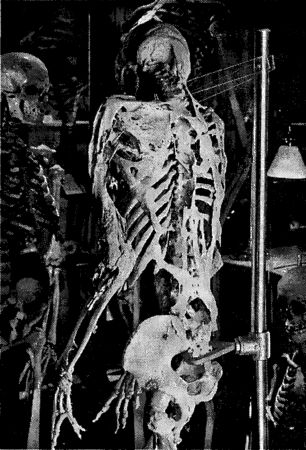
Как растут кости Скелет новорожденного младенца представляет собой произведение филигранной красоты и сложности; череп его по мягкости сравним с листом картона, но по толщине уступает ему, а бедренные кости тоненькие, как ствол карандаша. Когда ребенок вырастет и превратится во взрослого, все вышеупомянутые параметры изменятся. Бедренные кости в диаметре сравняются с хоккейной клюшкой и смогут, по крайней мере в большинстве случаев, выдерживать ее удары. Кости черепа по толщине сравняются с тарелкой и станут способны защитить мозг своего владельца, даже если он займется игрой в регби или станет участником другого, не менее любопытного действа, распространенного среди австралийских аборигенов, когда они, по заведенному обычаю, бьют друг друга по голове здоровенными палками.[129] Что же заставляет кость вырастать до нужного размера? В 1930 году молодой американский ученый Виктор Чендлер Твити попытался получить довольно-таки прямой ответ на этот вопрос. Следуя в русле немецкой школы "Механики развития" ("Entwicklungsmechanik"), Твити взялся изучать два вида саламандр: тигровую саламандру и пятнистую саламандру. Эти близкородственные животные различаются в одном, очень важном отношении: тигровые саламандры примерно вдвое больше пятнистых. Эксперименты, выполненные на них Твити, отличаются такой элегантностью, простотой и смелостью, что и спустя семьдесят лет фигурируют в большинстве учебников. Твити начал с того, что стал отрезать ноги у саламандр. Итальянский ученый Ладзаро Спаланцани из Скандиано еще в 1768 году установил, что у саламандр могут при необходимости отрастать ноги и хвост. С тех пор многие тысячи этих созданий ради науки лишались своих конечностей. Одна из неудачниц двадцать раз расставалась с ампутированной ногой – и всякий раз отращивала ее снова. По одному шутливому замечанию, которое распространено среди ученых, счастье состоит в том, чтобы найти удачный эксперимент и повторять его снова и снова. Твити, однако, был более изобретателен. Когда у саламандр зажили культи отрезанных конечностей и их ткани реорганизовались в зачатки новых конечностей, он снова взялся за нож и отрезал их. Затем он взял ампутированные почки конечностей одного вида и пересадил их на культи другого. Вопрос состоял в том, до какого размера будут расти чужеродные конечности. На то, по логике Твити, имелось две возможности. По мере того как пересаженные почки будут превращаться в ноги, они могут либо приобрести свойства хозяина, либо сохранить свои собственные. В первом случае почка конечностей пятнистой саламандры, пересаженная на культю тигровой, разовьется в мощную ногу, соответствующую размерам тигровой саламандры. Или, в ином случае, почка конечностей пятнистой саламандры просто вырастет в небольшую ногу, как это с ней обычно и происходит. В результате получится тигровая саламандра с тремя большими ногами и одной маленькой, пересаженной, короче говоря, кривобокая саламандра. Сам Твити ожидал, что чужеродные ноги вырастут столь же большими, как и обычные ноги саламандры-хозяина. В 1930-х годах уже было известно, что огромное влияние на процессы роста оказывают гормоны. Один из них, продуцируемый гипофизом, даже получил название гормона роста, и клиницисты говорили о людях с его избытком или дефицитом как о "гипофизарных" гигантах и карликах. Если тигровые саламандры были крупнее пятнистых, то происходило это наверняка потому, что первые имели больше гормона роста (или чего-то наподобие него), чем их более мелкие родственники. Чужеродные конечности должны реагировать на концентрацию гормонов хозяина в той же степени, что и обычные конечности, и, в соответствии с ней, становиться большими или маленькими. Контроль над ростом будет в этом смысле глобальным, когда все ткани должны подчиняться единому набору инструкций, циркулирующих по всему телу. Нет сомнений, что гормоны действительно играют роль, причем жизненно важную, в том, до какого размера вырастают саламандры, люди и, вероятно, вообще все животные. Красота эксперимента Твити заключалась тем не менее в другом: он показал, что как бы ни были важны гормоны, не они ответственны за различия между крупными и мелкими саламандрами. Против ожидания, его саламандры оказались кривобокими. Как будто пересаженная конечность неким непостижимо таинственным образом попросту знала, какого размера должна достичь, вне зависимости оттого, к чему ее прикрепили. Это был эксперимент, показавший первичность локального над глобальным, ибо любая нога саламандры несла в себе предсказание собственной судьбы.[130] В награду за эти эксперименты Твити стяжал впоследствии не слишком громкую славу. Более непосредственным откликом стала его поездка в Берлин в 1931 году. Он отправился работать в Институт кайзера Вильгельма, в лабораторию Отто Мангольда, мужа Хильды Прешельдт, прославившейся открытием организатора. Там он встретился с выдающимися биологами своего времени: Гансом Шпеманом, Рихардом Гольдшмидтом и Виктором Хамбургером, которые вместе создали приоритет Германии в области биологии развития. Ни исследования Твити в Институте кайзера Вильгельма, ни его дальнейшая карьера в Стэнфорде, где он сделался одним из наиболее любимых студентами профессоров, не представляют для нас особого интереса, в отличие от времени и страны, в которой он тогда оказался. В 400 километрах к югу, в Мюнхене, другой молодой ученый со схожими научными интересами, но совершенно иными моральными устоями, только что поступил на медицинский факультет. Это был Йозеф Менгеле. Освенцим, 1944 Тот, чье имя навсегда повисло мрачной тенью над исследованиями по генетике человека, происходил из семьи состоятельных баварских промышленников. Красивый, вежливый и интеллигентный, он отказался от службы в семейной фирме и вместо этого занялся изучением медицины и философии в Мюнхенском университете. Он был честолюбив и горел желанием сделать себе имя, первым в семье прославившись на научном поприще. К середине 30-х годов он переехал во Франкфурт, где его покровителем стал Отмар Фрайгерр фон Вершуер, глава еще одного Института кайзера Вильгельма, в котором, однако, занимались в основном антропологическими исследованиями. Диссертация, написанная там Менгеле в 1935 году, отражала присущую немецкой антропологии страсть к расовым классификациям и содержала измерения сотен челюстных костей для выявления расовых различий. Две более поздние научные статьи касались наследования некоторых телесных нарушений, таких как волчья пасть. Все эти работы суховаты, основаны на фактах и, в общем, довольно скучны. В них нет и намека на будущую карьеру молодого ученого. Менгеле приехал в Освенцим 30 мая 1943 года. Он отправился туда по настоянию своего ментора, фон Вершуера. Именно он убедил ученика воспользоваться теми, как он выразился, "необыкновенными исследовательскими возможностями", которые перед ним откроются. Ко времени прибытия Менгеле в концентрационный лагерь в нем содержалось чуть больше 100 тысяч заключенных, и машина смерти работала на полную мощность. Менгеле был лишь одним из представителей многочисленного медицинского персонала в Аушвиц-Биркенау, к тому же не занимавшим высоких постов. Но после войны люди, пережившие лагерь, будут вспоминать именно его. Он запомнится им своей физической красотой, всегда идеальным состоянием военной формы, очаровательными манерами и улыбкой. Они запомнят его благодаря непостижимому свойству его личности: он мог мило поговорить с ребенком, а потом отправить того в газовую камеру. Они запомнят его, потому что он казался вездесущим, и еще потому, что нередко он был первым немецким офицером, которого они встречали. Когда заключенные выходили из телячьих вагонов на платформу в Биркенау, они слышали, как он кричит "налево" или "направо". "Налево" означало немедленную смерть, "направо" – что им сохраняют жизнь, по крайней мере на время.[131] Среди тех, кого оставили в живых, была тридцатилетняя еврейская женщина по имени Элизабет Овиц. Вместе с братьями и сестрами она прибыла в Аушвиц-Биркенау ночью 18 мая 1944 года. Их привезли в вагоне для перевозки скота вместе с 84 другими людьми. Слабые и растерянные после долгого пути, Овицы стояли на платформе Биркенау под ослепительным светом прожекторов. Элизабет спросила одного узника, еврея-инженера из Вены, где они находятся. Он ответил: "Это могила Израиля" – и показал на дымящиеся трубы, которые возвышались над лагерем. Через сорок три года она напишет: "Теперь мы поняли, что все, о чем знали раньше, но пытались вытравить из своего сознания, происходит на самом деле". Элизабет и остальных членов семьи – группу в двенадцать человек – отогнали к краю платформы. Именно тогда они впервые увидели Менгеле. Восхищенно осматривая их, он заявил: "Теперь у меня есть работа на двадцать лет вперед; теперь у науки есть интересный объект для размышлений". Овицы были трансильванскими евреями. Их отец, Шимшон Исаак Овиц, был ученым человеком и известным раввином, который мог творить чудеса. У него была разновидность карликовости под названием псевдоахондроплазия, особенность которой состоит в том, что тело больного остается почти неизменным, а конечности вырастают короткими и искривленными. Равви Овица почитали за его мудрость и сострадание. Многие румынские евреи верили, что Бог, лишив его нормального роста, взамен наградил необычными и редкостными качествами. Амулеты с кусочками пергамента, на которых клубились завитки древнееврейского письма, изящно выписанные его рукой на раши[132] как говорили, обладали целительной силой. У равви Овица было десять детей, семеро из которых, включая Элизабет, были карликами. Это совпадает с диагнозом псевдоахондроплазии, которая вызывается доминантно наследуемой мутацией. 
Когда Элизабет исполнилось девять лет, ее отец внезапно умер. Молодая вдова, женщина весьма находчивая, сочла, что малый рост ее детей может быть использован для их же пользы. Она дала им музыкальное образование, и впоследствии они смогли организовать труппу. Даже в то время, когда Румыния и Венгрия уже оказались втянутыми в политическую орбиту нацистской Германии, семейство Овиц со своим "Джаз-оркестром лилипутов" все еще колесило по провинциальным городкам расколотых и нестабильных государств Центральной Европы. В мае 1942 года Элизабет Овиц, которой тогда исполнилось двадцать восемь лет, встретила молодого театрального менеджера по имени Йошко Московиц. Он был высокий, красивый и без памяти влюбился в нее. Он написал сестре, что встретил женщину хотя и маленького роста, но с лихвой одаренную талантом, мудростью и трудолюбием. Они поженились в ноябре того же года, но всего через десять дней после свадьбы Йошко с нашитой желтой звездой Давида на рукаве пиджака был отправлен в трудовой батальон. Супруги встретились снова лишь по окончании войны. Скрывая свое еврейское происхождение, Овицы продолжали гастролировать еще в течение двух лет, но в марте 1944 года немецкие войска оккупировали Венгрию, и во время последних и самых страшных погромов, прокатившихся по стране, их все же схватили. В Освенциме Элизабет вместе с братьями и сестрами держали в отдельной комнате, чтобы они не теснились в блоке вместе с пятьюстами его обитателями. Им разрешали также носить собственную одежду и давали достаточно пищи, чтобы выжить. В течение какого-то времени они смогли оставаться вместе как отдельная семья, и им даже удалось убедить Менгеле, что они являются родственниками еще одного семейства из их деревни. Они платили за свое выживание тем, что исполняли заглавные роли в нелепой и безумной программе экспериментальных исследований Менгеле. Впоследствии Элизабет Овиц напишет:
В этом, однако, Элизабет ошибалась. Менгеле подвергал пыткам множество других людей, в том числе немалое число близнецов, которых он в конечном итоге убивал и препарировал с единственной целью документально подтвердить сходство их внутренних органов. В течение семи месяцев Овицы балансировали между жизнью и смертью, оставаясь предметом одержимости Менгеле. Однажды, когда Менгеле неожиданно вошел в их отсек, самый младший в семье, Шимшон, которому едва исполнилось полтора года, неуверенно ковыляя, приблизился к нему. Менгеле взял ребенка на руки и мягко спросил, почему тот подошел к нему. "Он думает, что вы его отец". – "Я не его отец, – сказал Менгеле, – а всего лишь дядя". Тем не менее ребенок был крайне истощен от недоедания и постоянного забора крови. Менгеле демонстрировал Овицов высшим нацистским чинам. Он читал лекции о феномене карликовости, а в качестве иллюстрации выводил на сцену членов семьи Овиц, которые стояли там, обнаженные и дрожащие от холода и испуга. Эксперименты продолжались до октября 1944 года. И даже когда Третий рейх уже корчился в предсмертных муках, Менгеле не мог отказаться от своих маниакальных идей и пытался найти среди набора стеклянных глаз такие, которые по цвету подходили бы к карим глазам Элизабет. Как и во всех его поступках, причина этих действий остается загадкой. Освенцим был освобожден 27 января 1945 года. Для Элизабет и всех членов ее семьи приход советских войск означал отмену неизбежного смертного приговора. Почти все объекты опытов Менгеле подлежали уничтожению, как только эксперименты с ними заканчивались. В течение следующих четырех лет семейство странствовало по разрушенным городам Восточной и Центральной Европы. Реорганизовав труппу, Овицы исполняли мрачное танго, которое назвали танцем смерти[133]. Каждый вечер Элизабет в паре с одним из братьев танцевала партию Жизни, тогда как ее партнер изображал Смерть. В 1949 году семья эмигрировала в Израиль. Элизабет Овиц умерла в Хайфе в 1992 году.[134] Йозеф Менгеле так и не был предан суду за свои преступления и умер на бразильском пляже в 1979 году. Тормоза История семьи Овиц содержит в себе немало горькой иронии, но один из наиболее ярких ее примеров состоит в том, что Йозеф Менгеле был совершенно прав, когда считал этих людей удивительными. Лица с нарушениями наподобие псевдоахондроплазии действительно сообщают нам нечто важное о том, как кости вырастают до той длины, которая им положена, и каким будет наш рост. Менгеле не обнаружил, в чем причина карликовости, да его бессмысленные эксперименты никогда бы ему об этом и не рассказали. Но полстолетия спустя стало ясно, что обрубленные, искривленные и деформированные конечности, возникающие вследствие многих костных нарушений, говорят о явлении, открытом Виктором Твити: локальном контроле над ростом. Динамическая природа кости нигде не проявляется с такой очевидностью, как на концах длинных костей у ребенка. Каждый конец имеет область – ростовую пластинку, из которой осуществляется рост кости. В отличие от самой кости, заключенной в футляр из фосфата кальция, ростовые пластинки остаются мягкими и некальцифицированными. На рентгенограммах они выглядят как темные поперечные полоски, пересекающие белые концы каждой кости. Их можно наблюдать у ребенка на протяжении всего детства и отрочества: они постепенно уменьшаются в размерах и примерно к восемнадцати годам закрываются, знаменуя прекращение линейного роста. Каждая ростовая пластинка содержит сотни колонок хондроцитов, которые делятся и дифференцируются в четко согласованном порядке. Рождаясь на конце ростовой пластинки, наиболее удаленном от тела кости, они затем наполняются белком, из которого продуцируют вокруг себя хрящеобразный матрикс, после чего умирают. Остеобласты шагают по кладбищу хондроцитов, откладывают фосфат кальция и новые слои матрикса, вследствие чего кость с каждого конца растет все дальше и дальше. Псевдоахондроплазия, которой страдали Овицы, нарушает последовательность событий костеобразования. Мутация поражает ген, кодирующий один из белков, который входит в состав создаваемой хондроцитами хрящевой основы. Вместо того чтобы секретироваться клеткой, мутантный белок скапливается в хондроцитах, отравляя и убивая их задолго до положенного им срока. И хотя не все хондроциты погибают, воздействие этого нарушения столь велико, что рост резко замедляется. В результате возникают короткие, искривленные конечности, в то время как туловище и лицо практически не претерпевают никаких изменений.[135] Псевдоахондроплазия – лишь один из числа нескольких недугов, вызывающих появление очень коротких конечностей. Другим примером является собственно ахондроплазия – нарушение, с которым псевдоахондроплазию долго путали. От Птаха-Патайкоя, карликового божества юности, созидания и возрождения в Новом царстве Египта (1539-750 гг. до н.э.), до телевизионной рекламы газированных безалкогольных напитков, пожалуй, не найти другого такого часто встречающегося расстройства, которое столь широко использовалось бы в иконографии низкорослости.[136] Подобно одноименному недугу, ахондроплазия обусловлена недостатком хондроцитов, путешествующих по ростовой пластинке, но этот дефицит имеет совершенно иное происхождение. Ахондроплазия вызывается мутацией в рецепторе фибробластного фактора роста (FGF – fibroblast growth factors). FGF – это сигнальные молекулы, входящие в состав молекулярных часов, регулирующих рост конечности зародыша по продольной оси – от ближнего до дальнего ее конца. Однако после рождения FGF, вместо того чтобы способствовать росту конечности, тормозят его. Мы знаем об этом, потому что 99 процентов всех случаев ахондроплазии обусловлены мутацией, в результате которой одна аминокислота (глицин) в определенном локусе белковой последовательности FGFR3 (позиция 380) замещается другой (аргинином). Эта мутация обладает своеобразным свойством стимулировать повышенную активность молекулы FGFR3. Почти все мутации, которые описаны в этой книге, вызывают дефицит одного из белков, либо количественный, либо качественный, понижая степень его эффективности; нередко мутация служит причиной полного отсутствия белка. Если белок представляет собой сигнальную молекулу, как в случае с FGF, наблюдаемое нарушение будет следствием утраты некоего критического блока информации, который требуется клеткам. Мутация ахондроплазии, однако, отличается в том отношении, что заставляет рецептор передавать сигнал внутрь клетки даже при отсутствии связи с FGF. Вызываемый эффект подобен переключателю, который спонтанно включается, вместо того чтобы быть выключенным, и посылает часть нежелательной информации клеткам растущей конечности.[137] Если избыток сигнальных молекул FGF заставляет конечности необычно укорачиваться, значит, привычная роль FGF должна состоять в том, чтобы тормозить процесс роста конечности ребенка. Они выполняют эту функцию за счет ограничения скорости деления клеток в ростовой пластинке. Кости ребенка с ахондроплазией имеют ростовые пластинки, по размеру составляющие лишь малую толику того, какими они должны быть. По сравнению со здоровыми детьми в них содержится намного меньше делящихся хондроцитов и еще меньше тех, которые разрастаются и образуют хрящ.[138] Ахондроплазия – довольно-таки нестрашное нарушение. Однако избыток сигнальных FGF, в своей крайней форме, может вызывать ужасные последствия. В числе многих скелетов амстердамского Музея Фролика есть один, принадлежащий младенцу мужского пола, родившемуся мертвым где-то в начале 1800-х годов. При взгляде на скелет, ныне обозначенный номером М715, сразу становится ясно, что с ним что-то не так. Все до единого позвонки, ребра и тазовые кости ребенка усечены, искривлены или уплощены, а череп неимоверно увеличен. В своем грандиозном тератологическом трактате 1849 года Виллем Фролик описывает лоб ребенка как огромный шишковатый объект. Укороченные конечности и большая голова – оба эти симптома характерны для летальной скелетной дисплазии. Как явствует из самого названия, она приводит к смертельному исходу уже при рождении. 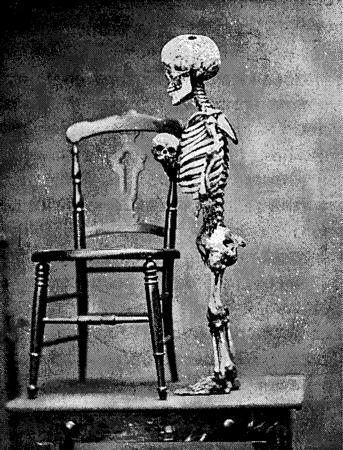
Летальная скелетная дисплазия также вызывается активирующими мутациями гена FGFR3, но намного более разрушительного свойства, чем те, которые ответственны за ахондроплазию. Причиняемый ими вред показывает, что семейство FGF контролирует рост не только конечностей, но и некоторых других частей скелета, таких, например, как череп. Слегка напоминающие купол черепа многих карликов с ахондроплазией наводят на мысль о том, что их заболевание является ослабленной версией смертельного недуга. Если ребенок унаследует две копии ахондропластической мутации (имея, к примеру, двух родителей-ахондропластиков), то он умрет вскоре после рождения со всеми симптомами летальной скелетной дисплазии.[139] FGF – это, должно быть, лишь одна молекула в числе многих, ограничивающих рост той или иной части скелета. Каждый орган должен имеет механизмы, диктующие ему, когда нужно прекращать рост, причем многие из этих механизмов уникальны для конкретного органа. Едва ли существует хоть одна часть тела, которая бы не затормаживала или не ускоряла свой рост в результате того или иного генетического расстройства. Некоторые мутации заставляют детей рождаться с такими большими языками, что они не влезают в рот; в результате других – кишечник не помещается внутри брюшной полости.[140] Даже у мышц есть свои собственные механизмы регуляции роста. Бельгийские коровы специальной мясной породы замечательны тем, что имеют на треть больше мышц по сравнению с обычными коровами. Их бока напоминают ляжки олимпийских чемпионов-тяжелоатлетов. У них отсутствует белок, называемый миостатином (родственный, так уж случилось, молекулам BMP), который приказывает мышцам прекращать рост. У мышей, дефектных по миостатину, мышечная масса в два-три раза больше обычной, но цена этого избыточного роста оплачивается за счет его дефицита в других местах, поскольку внутренние органы у них вырастают меньше нормы. Наверняка существуют и люди с дефектами миостатина, но достоверные свидетельства о них отсутствуют. Возможно, экстра-мускулатура остается незамеченной, а если ее и замечают, то не считают причиной для беспокойства.[141] Обновление Ген, ответственный за axoндpоплазию, кодирует некоторую часть матрикса, который продуцируют вокруг себя хондроциты. Но это лишь малая его доля. Действительно, мыши, у которых этот белок полностью удален путем генетического инженеринга, вообще как будто не страдают от вредных последствий таких манипуляций. Можно вообще в первую очередь задаться вопросом, какова роль этого белка в матриксе. Далеко не так обстоят дела с остальным матриксом. Большая часть хряща состоит из коллагена. У людей насчитывается до пятнадцати различных типов коллагена, которые составляют около четверти всего количества белка в организме. Разновидности коллагена обнаруживаются в соединительной ткани и коже. Они соединяют вместе наши клетки. И именно они в значительной степени обеспечивают свойственные кости гибкость и силу. Мутации, повреждающие костные коллагены, вызывают нарушение, носящее название несовершенного остеогенеза (osteogenesis imperfecta). Существует по крайней мере четыре формы, объединяемые под этим диагнозом, причем некоторые из них заканчиваются летальным исходом в младенчестве. Наиболее характерный симптом этого генетического расстройства – чрезвычайная хрупкость костей у его жертв. По этой причине его также часто именуют болезнью стеклянных костей. Мутации оказывают свое разрушающее воздействие из-за иерархической природы, которой подчиняется структура коллагенов. Любой коллагеновый белок образован тремя пептидами – цепочками аминокислот, свернутыми вместе в тройную спираль. Тройные спирали, в свой черед, группируются вместе, образуя огромные фибриллы, или волокна, которые, сплетаясь вместе, определяют строение соединительной ткани или хряща. Каждый пептид кодируется отдельным геном, но один-единственный мутантный ген может разрушить любое число тройных спиралей и, следовательно, любое число фибрилл, а значит, и костей.[142] От несовершенного остеогенеза страдали французский художник Ашиль Амперер (1829-1898), портрет которого нарисовал Сезанн, и французский джазовый пианист Мишель Петруччиани (1962-1999). Эти артистические ассоциации придали болезни, по крайней мере во Франции, псевдоромантический ореол (на ум приходит "человек со стеклянными костями" из фильма Жан-Пьера Жене "Амели"). Реальность куда более прозаична. Дети с несовершенным остеогенезом часто страдают небольшими костными переломами, о которых родители вообще не подозревают. Когда же, после более серьезного перелома, дети наконец попадают в больницу, рентгеновское обследование выявляет большое количество ранее поломанных и заживших костей. На этой почве часто возникают подозрения, что родители жестоко обращаются с детьми. В Соединенных Штатах больные дети попадали под опеку сверхретивых социальных работников, а некоторые родители подчас отправлялись за решетку. 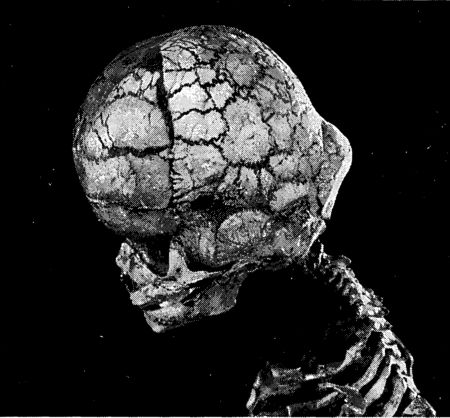
Даже после того как ростовые пластинки закрываются и прекращается рост, скелет не знает покоя. Внутреннее содержимое большинства костей у взрослого человека полностью замещается каждые три или четыре года, в то время как наружные, более твердые части подлежат замене примерно раз в десятилетие. Этот процесс разрушения и обновления осуществляется на основе союза между остеобластами и другими клетками, которые постоянно поглощают скелет, откусывая маленькие кусочки от его основы и высвобождая тем самым необходимые составные части, наподобие множества резцов. Это остеокласты – гигантские клетки, которые прикрепляются к костным фрагментам и растворяют их с помощью поедающих белок ферментов и соляной кислоты. Кости строятся с помощью остеобластов, но свои окончательные формы они принимают при участии остеокластов, так как именно эти клетки высекают протоки, каналы и полости для прохождения нервов и кровеносных сосудов и размещения костного мозга.[143] Существует немало способов, чтобы сдвинуть равновесие между ростом и разрушением, которое характерно для каждой кости. Избыток костного вещества может быть следствием избытка остеобластов или нехватки остеокластов. Остеопетроз, в буквальном переводе – каменные кости[144], – заболевание, связанное с остеокластами.[145] Оно противоположно намного более известному остеопорозу, которое становится настоящим бичом для женщин постклимактерического возраста.[146] Состояние, при котором кости делаются как камни, может быть летальным. Существует особенно тяжелый вариант этого расстройства, поражающего детей и убивающего их до достижения двадцатилетнего возраста. Они часто умирают от инфекционных заболеваний, так как кость аккумулируется в полости, служащей для производства костного мозга, который является одним из основных источников клеток иммунной системы. Кажется парадоксом, что кости людей, больных остеопетрозом, также имеют склонность довольно легко ломаться. Это, вероятно, происходит вследствие нарушения архитектоники кости. И когда переломы случаются, их заживление происходит нелегко, так как остеокласты, в числе прочих своих функций, выравнивают зазубренные края костей, если уж мы их поломали. По мнению некоторых, остеопетроз, хотя и в довольно мягкой форме, был причиной маленького роста Анри де Тулуз-Лотрека. Это лишь один из нескольких диагнозов, которые задним числом пытаются поставить французскому художнику. Среди других – ахондроплазия и несовершенный остеогенез. Ни один из этих диагнозов не является достаточно убедительным. Существует такое огромное количество костных нарушений, а симптомы их столь разнообразны и трудноотличимы, что их легко перепутать, тем более если все, что мы знаем о пациенте, – это сведения из его биографии, пачка фотографий и серия автопортретов, по большей части карикатурных. И все же поиски "болезни Лотрека" продолжаются. Притягательность этого занятия, по крайней мере для французских врачей, отчасти объясняется тем, что художник был потомком одного из самых знатных семейств Франции – графского дома Тулуз-Лотреков, династии непокорных аристократов Юга, которые в то или другое время правили большей частью Руерга, Прованса и Лангедока, грабили Иерусалим, не чурались ереси, бывали отлучены папой от церкви (в десяти отдельных случаях) и в XIII веке испытали на себе ярость и военную мощь французской короны. Но сверх всего этого за стремлением найти причину болезни Анри Тулуз-Лотрека стоит убеждение, что этот одаренный художник сделал собственное уродство частью своего искусства. В этом, возможно, что-то есть. Пройдитесь по залам Музея д'Орсэ в Париже или музея в Альби, неподалеку от самой Тулузы, полностью посвященного его работам, и вас поразят ноздри на его полотнах. Просматривая картину за картиной – портреты танцовщицы Гулю, актрисы Иветт Гильбер, светской львицы Мей Мильтон или многих других, неизвестных представительниц парижского полусвета, которые населяют живопись Лотрека, вы в первую очередь видите ноздри – их зияющие, темные провалы. Этот ракурс едва ли можно назвать лестным для его моделей, но, возможно, именно такими видел их художник, поскольку был довольно невысоким. К моменту окончания роста длина его тела составляла менее 150 сантиметров. Критики также утверждают, что болезнь Лотрека оказала и другое, более незаметное воздействие на его искусство: после 1893 года в нем обнаруживается четкая тенденция удалять у моделей ноги, так чтобы в раме оставались только головы и торсы. Этот прием позволял исключить ту часть анатомии, о которой он сам предпочел бы забыть. 
Ноги Лотрека причиняли ему много страданий. У него было как будто вполне счастливое детство, но когда ему исполнилось семь, мать повезла его в Лурд, где надеялась найти исцеление от каких-то проблем с ногами, довольно смутно описанных. Он был скован, неуклюж и часто падал. В школу он проходил всего один год, после чего его забрали домой, так как потасовки на школьном дворе были явно не для него. К десяти годам он начал жаловаться на постоянные сильные боли в ногах и бедрах; в тринадцать лет незначительное падение привело к переломам обеих бедренных костей, которые, если судить по тому промежутку времени, когда ему пришлось передвигаться, опираясь на две трости, срослись только через полгода. Он будет пользоваться тростью на протяжении практически всей своей взрослой жизни, и его друзья, безусловно, верили, что он преодолевает любое расстояние с неохотой и с большим трудом. По мере роста необычные изменения произошли и в его внешности. Он был хорошеньким малышом, затем красивым мальчиком, но впоследствии у него отвисла нижняя губа, стала течь слюна, речь сделалась непонятной и отрывистой, появилась шепелявость, а зубы сгнили еще в отрочестве. Все это отличало его от родителей, которые были очень хороши собой. Он стеснялся своего вида, всю жизнь носил бороду и никогда не улыбался перед камерой. Многие критики утверждают, что именно своеобразная ненависть к собственному телу заставляла его искать и изображать то, что было наиболее некрасивым и неприятным в его milieu[147]. Но Париж fin-de-siecle[148] вполне мог быть некрасивым и неприятным местом. Однажды ночью у "Максима", где Лотрек делал молниеносные карикатурные зарисовки своих соседей, один из них обратился к нему, заметив, что тот захромал прочь. "Месье, вы забыли свою трость," – сказал он, указывая на огрызок карандаша, оставленный на столе. В другой раз Иветт Гильбер, разглядывая один из своих многочисленных написанных им портретов, заявила: "Действительно, Лотрек, – ты гений безобразного". Он ответил: "Да, так оно и есть". Лотрек, как считают, страдал разновидностью остеопетроза, так называемым пикнодизостозом, который вызывается дефицитом фермента, используемого остеокластами для растворения белкового матрикса кости. В зрелом возрасте активность этого фермента частично подавляется гормонами. Именно снижение концентрации эстрогена у женщин постклимактерического возраста, а следовательно, ненужная активность фермента являются причиной остеопороза. Диагноз пикнодизостоза был поставлен Лотреку в 1962 году двумя французскими врачами Пьером Марото и Морисом Лами, но их мнение не осталось неоспоренным. Джулия Фрей, автор последней биографии Лотрека, признает, что по крайней мере часть симптомов согласуется с этим диагнозом, но в то же время отмечает, что многие признаки для него совсем нехарактерны. У больных пикнодизостозом, как правило, мягкие головы, наподобие голов "бескостных" капских малайцев; в отношении Лотрека таких свидетельств нет, его голова была вполне крепкой. Каково бы ни было заболевание Лотрека, по-видимому, он разделял его с другими членами семьи. К 1864 году, когда родился Анри Мари Раймон граф де Тулуз-Лотрек Монфа, его семья, по-прежнему богатая, характеризовалась высокой степенью близкородственного скрещивания (инбредностью). Наполеоновская отмена права первородства подталкивала и без того резко сократившуюся французскую аристократию сохранять то богатство, которое еще оставалось в семьях, с помощью простого приема – заключения браков только в пределах дома. Родители Анри были двоюродными братом и сестрой, равно как и его дядя с тетей. Все вместе они произвели на свет шестнадцать детей, четверо из которых, включая Анри, были карликами, причем трое других значительно больше отставали в росте, чем он. На самом деле, весьма вероятно, что по меньшей мере некоторые из ныне живущих членов этого благородного клана все еще являются носителями мутации, хотя действие ее, скорее всего, не проявляется по той причине, что практика кровосмесительных браков прекращена. У самого Лотрека не было сомнений по поводу истинной причины его заболевания. Как-то ночью, когда он находился в одном из своих любимых прибежищ – Ирландском и Американском баре на Монмартре, две женщины заспорили по поводу несчастной собаки, чьи ноги тряслись от дисплазии бедренных суставов. Владелица собаки соглашалась, что та не отличается красотой, но тем не менее настаивала на ее чистопородности. "Ты что, шутишь, – у этой собаки и родословная? Да ты погляди на ее гадкий мех и искривленные ноги, – смеялась подружка. – На нее просто жалко смотреть". – "Ты явно в этом ничего не понимаешь, – ответила хозяйка собаки и повернулась к Анри, который сидел рядом с ней. – Скажите же ей, месье, что моя собака вполне может быть уродливой, но с хорошей родословной". Анри слез с высокого стула у стойки бара и, выпрямившись на все 150 сантиметров своего роста, взмахнул перепачканной углем рукой в знак приветствия, пробурчав: "Это вы у меня спрашиваете?"[149] Примечания:1 О монстре из Равенны писали многие. См. у Ландуччи (Landucci, 1542; 1927) стр. 249-250 описание глазами современника. Жан Сеар (Jean Ceard) обсуждает эволюцию монстра в издании Паре "О монстрах" (Pare, 1573, 1971), стр. 153-155; Никколи (Niccoli, 1990) пишет о его политическом значении; см. также Fischer, 1991, p. 54-56; Daston and Park, 1998, p. 177-182. Я полагаю, что у монстра был синдром Робертса (268300), но другие ученые (Walton et al., 1993; Martinez-Frias, 1993) считают, что это была циклопия, сиреномелия и даже гидроцефалия. Все это не более чем догадки, и тот диагноз, который вы выберете, зависит прежде всего от описания монстра, которым вы воспользуетесь, и от того, какие из множества странных его черт вам покажутся более реальными. — прим. авт. 12 Эйр-Уокер и Кейтли (Eyre-Walker and Keightley, 1999) подсчитали скорость образования вредоносных мутаций у человека. Их результаты согласуются с таковыми других авторов (Cargill et al., 1999; Stephens et al., 2001), полученными иным способом. Кроу (Crow, 2000) обсуждает воздействие новейших (novel), вновь возникших мутаций на приспособленность организма. — прим. авт. 13 Рассказы современников о Рите и Кристине Пароди приводят многие авторы (Anon, 1829 a, b, c; Saint-Ange, 1830; Danerow, 1830). Более поздние работы на эту тему: Thompson, 1930, 1996; Bondeson, 2000. — прим. авт. 14 Главный анатомический труд о Рите и Кристине – это монография Серра (Serres, 1832). Э. Жоффруа Сент-Илер (Е. Geoffroy Saint-Hilaire, 1829) посвятил девочкам небольшую статью, а И. Жоффруа Сент-Илер (I. Geoffroy Saint-Hilaire, 1832-1837) описал их на стр. 161-174 в 3-м томе своего сводного труда по тератологии. — прим. авт. 122 Piesang – банан (африкаанс). 123 Об Арнольде и его потомках пишут многие авторы (Jackson, 1951; Ramesar et al., 1996). Их заболевание называется ключично-черепной дисплазией (119600). Оно вызывается доминантным гаплонедостаточным аллелем в транскрипционном факторе дифференцировки остеобластов – CBFA1 (600211). Об идентификации мутации, ее функции у мышей, описании хромосомного нарушения см. у ряда авторов (Komori et al., 1997; Mundlos et al., 1997; Mundlos, 1999). Одна из небольших загадок, связанных с этим нарушением, состоит в том, что в Южной Африке отсутствуют дети – явные гомозиготы по этому признаку. При таком большом числе носителей в пределах маленького сообщества два носителя должны были бы в конце концов неизбежно вступить в брак. Если мутация действует у людей таким же образом, как и у мышей (а для подтверждения этого есть множество доказательств), тогда 25% детей от такого брака должны были бы рождаться мертвыми и полностью лишенными костей (у половины кости отсутствовали бы частично, а одна четверть оказалась бы нормальной). — прим. авт. 124 Дизостоз ключично-черепной (d. claviculocranialis; син.: Д. клейдокраниальный, Шейтхауэра-Мари-Сентона синдром) – характеризующийся незаращением родничков черепа, брахицефалией, гипоплазией лицевых костей (главным образом верхней челюсти), полным или частичным недоразвитием ключиц. — прим. перев. 125 Для более подробного ознакомления с процессами роста костей см. работу: Olsen et al., 2000. То, что сказано здесь о роли молекул BMP в образовании конденсатов, носит противоречивый характер; полученные на мышах доказательства скорее свидетельствуют о значении BMP в процессе паттернинга, чем в дифференциации остеобластов и хондроцитов или в образовании конденсатов (Wagner and Karsenty, 2001). Я полагаю, что это происходит благодаря избыточности молекул BMP. — прим. авт. 126 Склеростоз (269500) вызывается рецессивными мутациями секретируемого белка склеростина (605740). Вплоть до недавнего времени считалось, что семьи людей из Южной Африки (все – африканеры), семьи из Баии, Бразилия, а также голландские семьи с похожим нарушением, называемом болезнью Ван Бучема, или гиперостозом кортикальным генерализованным (hyperostosis corticalis generalisata) (239100), все связаны родственными узами, хотя и отдаленными. Однако теперь доказано, что в семьях африканеров, жителей Баии и Голландии присутствуют разные мутации, произошедшие либо в гене SOST, либо около него, так что родственниками эти люди быть никак не могут, и наличие сходного заболевания во всех трех популяциях является простым совпадением (Brunkow et al., 2001; Balemans et al., 2001). — прим. авт. 127 Болезнь сросшихся пальцев, или синдром проксимального симфалангизма (185800), вызывается доминантной гаплонедостаточной мутацией в ноггине (602991) (Gong et al., 1999). О нулевых мутациях у мышей см.: Brunet et al., 1998. — прим. авт. 128 Прогрессирующая окостеняющая фибродисплазия (fibrodisplasia ossificans progressive), или ПОФ (135100) вызывается доминантными мутациями неизвестного гена. В 2001 г. группа французских исследователей сообщила об открытии мутаций ноггина у больных ПОФ (Semonin et al., 2001), однако подтвердить эти данные пока не удалось (Cohen, 2002). История болезни Гарри Истлэка описана Уорденом (Worden, 2002). Хороший обзор клинической картины ПОФ, пораженных ею людей и поисков средств для излечения болезни дается в: Maeder, 1998. — прим. авт. 129 Бейкер (Baker, 1974) отмечает, что толщина черепной коробки у большинства взрослых составляет около 5 мм; у австралийских аборигенов эта цифра может доходить до 10 мм. Ритуал битья палками по голове вкратце описывает Кон (Kohn, 1995). — прим. авт. 130 Описание опытов Виктора Твити см. в двух работах (Twitty and Schwind, 1931; Twitty, 1966). Эти эксперименты были начаты по инициативе выдающегося специалиста по биологии развития Росса Харрисона и выполнены в его лаборатории в Йельском университете (Harrison, 1924). Описание процесса регенерации конечностей у саламандр см.: Brockes, 1998. — прим. авт. 131 Биография Менгеле приведена в работе: Posner and Ware, 1986. — прим. авт. 132 Шрифт Раши, названный по имени рабби Шломо Ицхака (аббревиатура: Раши) – известного комментатора Торы, Талмуда и Библии (1040-1105). — прим. перев. 133 Танец смерти – широко распространенная тема средневековых изображений, в частности фресок на стенах храмов в Западной Европе. — прим. перев. 134 Этот рассказ отчасти основан на воспоминаниях Элизабет Овиц (Moskovitz, 1987), которые послужили также источником приведенных цитат. Однако тщательное изучение истории членов семьи и их пребывания в Аушвиц-Биркенау (Koren and Negev, 2003) выявило некоторые неточности в этих воспоминаниях. Детальное описание медицинских экспериментов, проводимых на людях в Третьем рейхе, можно найти: Lifton, 1986. — прим. авт. 135 Псевдоахондроплазия (177170) вызывается доминантными мутациями "утраты функции" в гене хрящевого олигомерного матричного белка гена (600310) (Briggs and Chapman, 2002). То, что Овицы страдали именно этим заболеванием, а не ахондроплазией (как об этом часто пишут), доказывают Корен и Негев (Koren and Negev, 2003). Этот диагноз хорошо согласуется с их привлекательными лицами. — прим. авт. 136 В ряде работ дается исторический обзор иконографии карликовости (Dasen, 1993, 1994; Aterman, 1999). — прим. авт. 137 Ахондроплазия (100800) вызывается доминантными мутациями утраты функции в рецепторе 3 фибробластного фактора роста (FGFR3) (134934) (Rousseau et al., 1994; Bonaventure et al., 1996). — прим. авт. 138 О той роли, которую играют гены FGF в росте конечностей, сообщают многие исследователи (Naski et al., 1996, 1998; Chen et al., 2001). Колвин с соавторами (Colvin et al., 1996) изучали мышь с нокаутом FGFR3; Де Лука и Барон (De Luca and Baron, 1999) приводят обзор функций FGFR3. — прим. авт. 139 Летальная скелетная дисплазия (187600) вызывается тяжелыми доминантными мутациями утраты функций в FGFR3 (Rousseau et al., 1995; Tavormina et al., 1995). Экспонаты Музея Фролика с летальной скелетной дисплазией описывают: Oostra et al., 1998 b. — прим. авт. 140 Серьезный обзор синдромов, связанных с избыточным ростом, приводит Коэн (Cohen, 1989). — прим. авт. 141 О мутациях миостатина (601788) у мышей см. в работе: McPherron et al., 1997; у крупного рогатого скота: McPherron and Lee, 1998. Изначально мутация миостатина произошла естественным путем на одной фламандской ферме, и поэтому мясо бельгийских коров однозначно идет на гамбургеры. Если бы то же самое животное было получено путем инжиниринга компанией "Монсанто", широкая публика, с подозрением относящаяся к "генетически модифицированным продуктам", его бы наверняка отвергла. — прим. авт. 142 Существует несколько разновидностей несовершенного остеогенеза (osteogenesis imperfecta). Наиболее распространенная из них не приводит к летальному эффекту при рождении и носит название "несовершенный остеогенез 1-го типа" (166200). Это нарушение вызывается доминантными гаплонедостаточными мутациями ("утраты функции") в генах коллагена 1A2 и 1A1 (120150; 120160) (Olsen et al., 2000). — прим. авт. 143 Обзорную информацию о функциях и спецификации остеокластов см.: Gunter and Schinke, 2000. — прим. авт. 144 Остеопетроз (от греч. petra – камень), или болезнь Альберс-Шенберга, или мраморная болезнь – врожденное заболевание, характеризующееся злокачественным остеосклерозом с ломкостью костей и анемией. — прим. перев. 145 Злокачественный аутосомно-рецессивный остеопетроз (259700) вызывается рецессивными мутациями генов, кодирующих часть вакуолярного протонного насоса, необходимого для транспортировки соляной кислоты (Kornak et al., 2000). — прим. авт. 146 О соотношении между остеопетрозом и остеопорозом см.: Lazner et al., 1999. — прим. авт. 147 Окружение (фр.). 148 Конец века (фр.). 149 Биографические материалы и курьезные случаи из жизни художника взяты по большей части из авторитетного труда Джулии Фрей (Frey, 1994). Марото и Лами (Maroteaux and Lamy, 1965) пересматривают прежние диагнозы и считают, что у Лотрека был пикнодизостоз. Обмен мнениями по поводу диагностики заболевания см.: Frey, 1995 a, b и Maroteaux, 1995. Пикнодизостоз (265800) вызывается рецессивными мутациями в гене катепсин K (601105) (Celb et al., 1996). — прим. авт. |
|
|||
|
Главная | Контакты | Прислать материал | Добавить в избранное | Сообщить об ошибке |
||||
|
|
||||